
为规范医疗器械临床试验检查工作,统一检查范围和判定标准,提高医疗器械临床试验项目检查质量,国家药监局组织修订《医疗器械临床试验项目检查要点及判定原则》。新版《医疗器械临床试验项目检查要点及判定原则(2025年第22号)》将于2025年5月1日施行。
参考法规
国家药监局关于发布医疗器械临床试验项目检查要点及判定原则的公告(2025年第22号)
国家药监局综合司关于印发医疗器械临床试验检查要点及判定原则的通知(2018年)
医疗器械临床试验现场检查要点(2016年)
本文根据2016版(IVD部分)、2018版本(医疗器械部分)及2025新版法规对比,总结出新旧法规变化要点,帮助大家快速掌握关键变化。
01
文件结构与核心差异
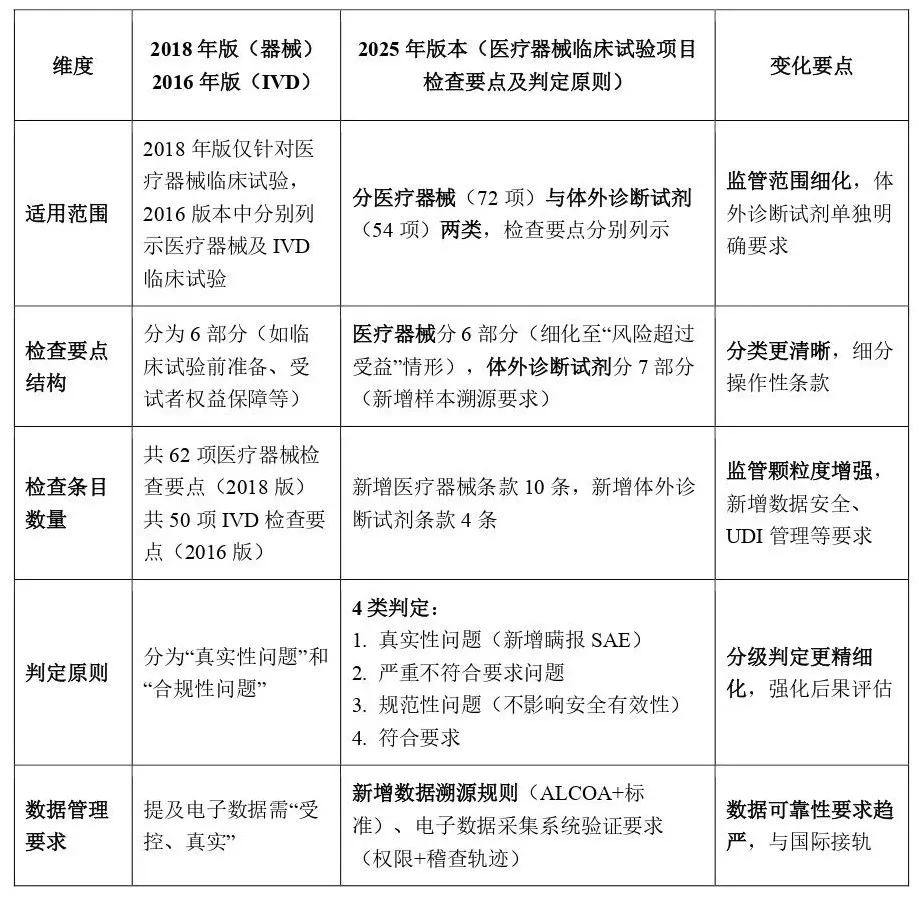
02
重点要求升级与监管趋势
• 医疗器械临床试验要求
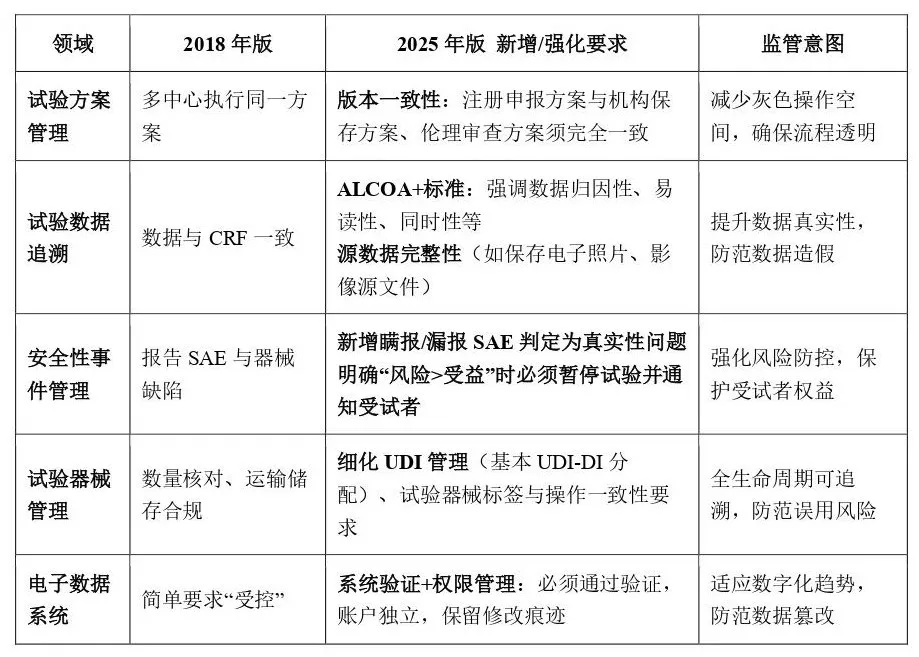
• 体外诊断试剂(IVD)临床试验要求
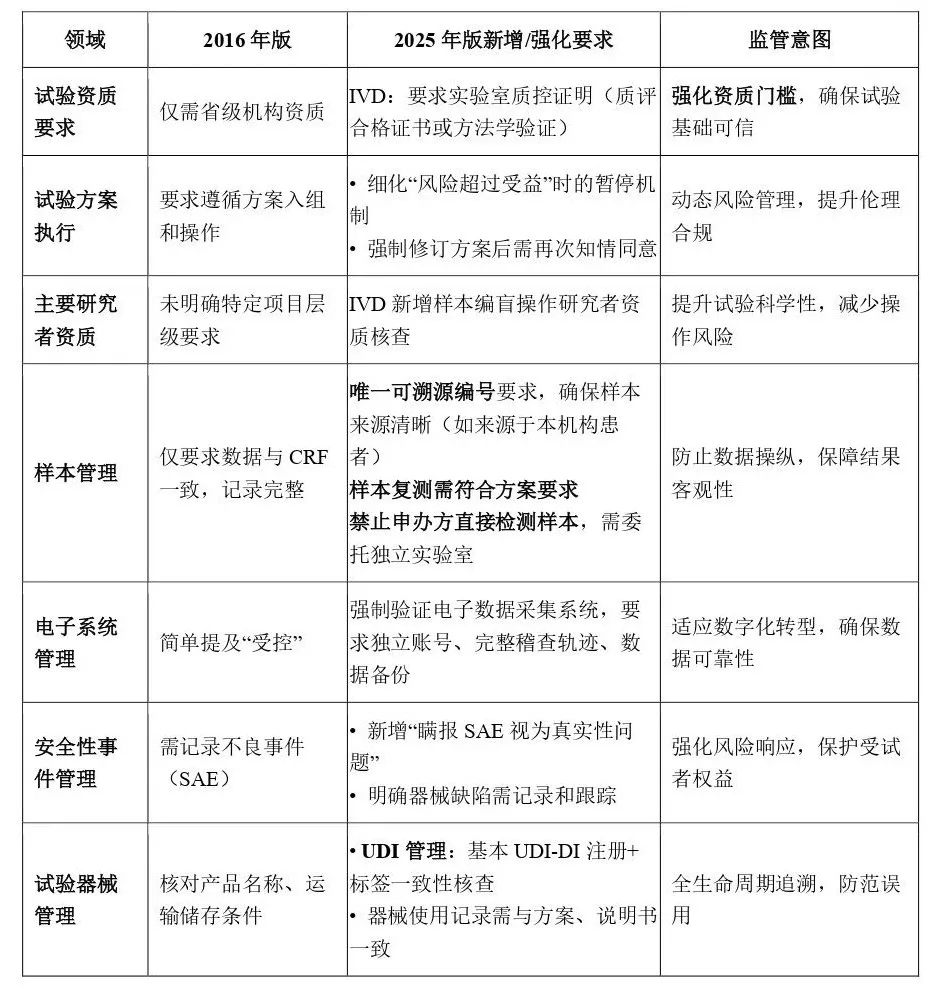
03
判定原则升级与监管趋势
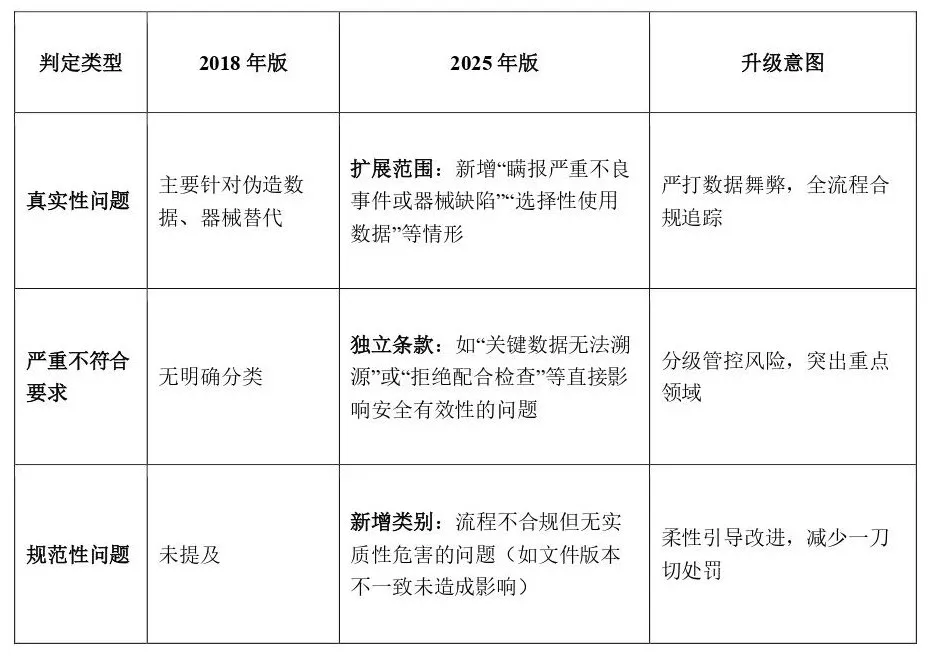
04
监管趋势总结
• 引入ALCOA+数据标准(欧盟要求)、UDI管理(全球监管趋势),推动中国临床试验数据与国际互认。
• 细化体外诊断试剂规则,向欧盟IVDR看齐,强化样本与实验室管理。
• 判定原则分级(真实性问题→严重问题→规范性→符合要求),区分故意造假与流程瑕疵,提升执法精准度。
• 增设安全性应急响应(如“风险>受益时立即暂停试验”),强化受试者保护。
数字化与透明度。
• 强制电子数据系统验证与轨迹追踪,适应远程监查趋势。
• 注册申报资料与机构内部资料一致性核查,倒逼企业规范化管理。
• 细化研究人员资质(如创新器械主要研究者需高职称+3项试验经验),提升试验科学性。
• IVD特殊监管:样本管理、实验室资质要求显化,确保检测结果客观性。
05
应对策略
• 建立符合ALCOA+的数据管理系统,完善电子数据稽查轨迹。
• 加强IVD样本全流程溯源,实施样本唯一编码,选择独立检测实验室并留存资质证明。
• 开展UDI编码培训,确保标签与数据库同步更新。
• 升级伦理委员会功能,增加SAE与方案偏离的实时监控能力。
• 制定分中心数据一致性核查SOP,定期交叉核验。
• 提前模拟检查中“严重不符合要求”场景(如数据库锁定的合规性)。
• 对研究者开展分级判定案例培训,减少规范性问题。
当前,新版法规的发布,标志着医疗器械临床试验监管迈入精准化、技术化、全周期化的新阶段。从数据管理(ALCOA+原则)到IVD样本全流程追溯,从研究者资质升级到SAE跨部门闭环报告,每一项变化都直指行业痛点,旨在筑牢医疗器械安全有效的基石。
对企业而言,合规已不仅是“底线要求”,更是核心竞争力。唯有主动拥抱变革,才能在严监管时代抢占先机。


